The COVID-19 pandemic has caused unprecedented public health and socioeconomic crises around the globe. The causative agent, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), spreads mainly through the respiratory system and, like many other viruses, keeps evolving into new viral variants with distinct characteristics.
The emergence of new and more infectious SARS-CoV-2 variants poses severe risks in the fight against the ongoing COVID-19 pandemic. Therefore, understanding the evolution of the virus and origin of the variants is critical for effectively controlling viral transmission as well as the global pandemic.
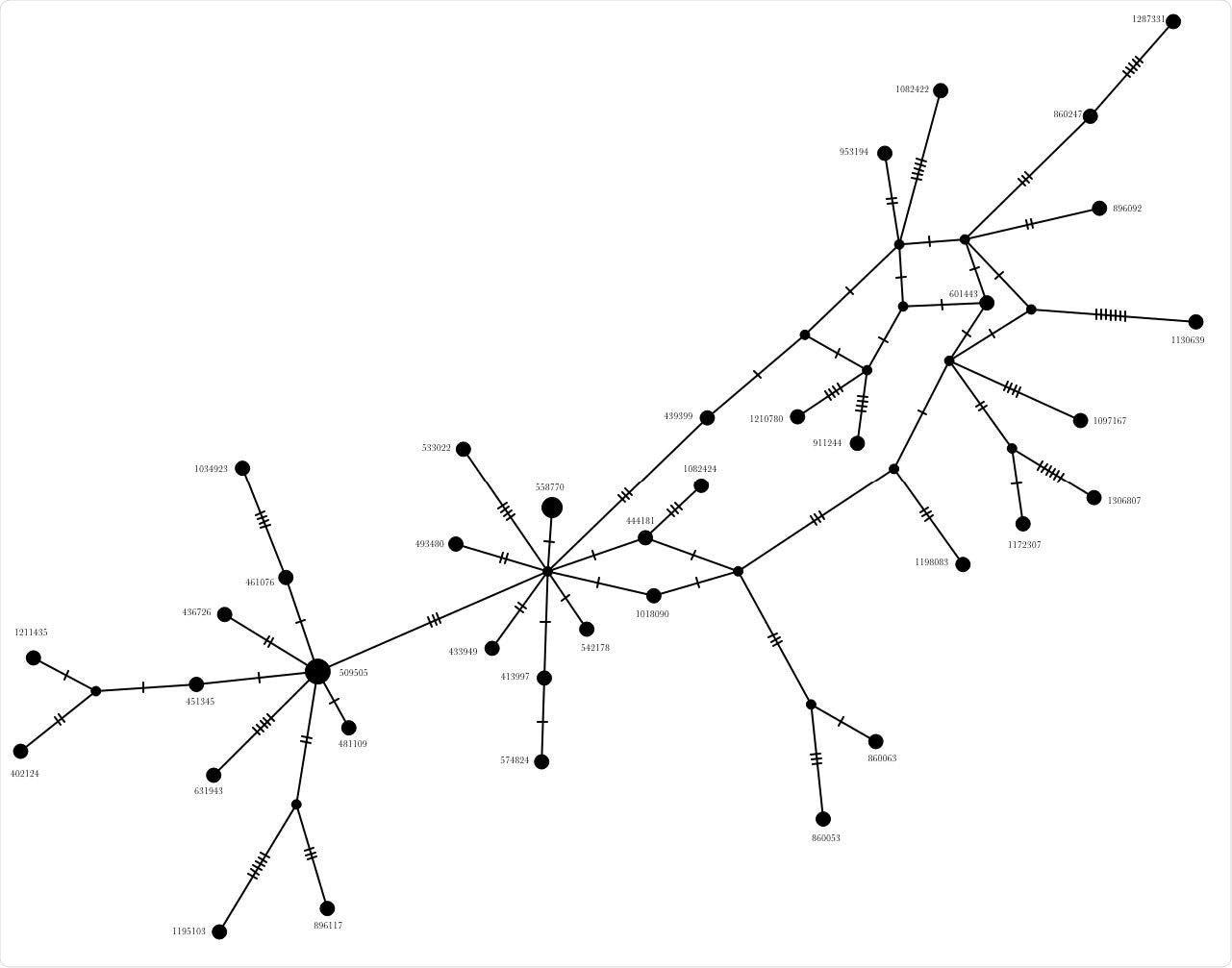
Using phylogenetic network approach to analyze the origin of the B.1.1.7 variant
To date, the study of virus evolution has been dominated by phylogenetic tree analysis. However, this method does not adequately answer a number of important questions.
Researchers from the U.S. recently used the phylogenetic network approach to analyze the origin of the VOC202012/01 (alpha) or the PANGO Lineage B.1.1.7 variant, which was first reported in U.K. in 2020 and soon spread to other countries. This study is published on the bioRxiv* preprint server while the paper undergoes peer review.
The alpha variant has been classified as a Variant of Concern and has led to a surge in COVID-19 cases and deaths in many countries. Studies show that the variant classified as PANGO lineage B.1.1.7 is more infectious compared to other strains of the virus in circulation in humans. The emergence and rapid spread of this and other variants adversely impact the fight against the COVID-19 pandemic both with respect to detection and the efficacy of currently available vaccines.
Thus, gaining a better understanding of how the U.K. variant could provide critical information about the processes behind the formation of new SARS-CoV-2 variants and help devise better strategies for controlling the pandemic.
Results show that the alpha variant could be a result of recombination of previously circulating variants
The multiple network analyses used different methods and consistently revealed that the B.1.1.7 variant resulted from recombination of pre-existing virus strains and not stepwise mutations in a linear order, as is commonly assumed. This highlights a key but largely ignored mechanism in SARS-CoV-2 evolution so far.
It is also suspected that other recently emerged variants such as the ones first reported in South Africa, India, and Brazil might also have been a result of the recombination of existing variants.
This new research demonstrates the competitive advantages of phylogenetic network analysis over phylogenetic tree analysis in the evolutionary study of the SARS-CoV-2 virus and its variants.
Despite phylogenetic tree analyses being relatively easy to conduct with well-established approaches resulting in more straightforward to understand results, unfortunately, the approach is not suitable to study the evolution of SARS-CoV-2 viruses for the following reasons.
For starters, phylogenetic tree construction places all existing sequences at the tips of the tree and artificially prevents any existing sequence from being an ancestor of some other sequence. Because the SARS-CoV-2 virus strains have a very short evolutionary history, some ancestral sequences that gave rise to other sequences might still be in circulation in human populations, and might have been sampled by researchers.
Secondly, bifurcating phylogenetic trees rule out the possibility of one ancestral sequence giving rise to more than two descendant lineages.
The latter possibility may well have been present in the evolution of SARS-CoV-2 viruses.
Finally, phylogenetic trees assume no recombination between or among any ancestral sequences.
Several RNA viruses, including HIV, polioviruses, bromoviridae, influenza, Western equine encephalitis viruses, and coronaviruses, can recombine through template switching.
In light of the above, phylogenetic network analysis would be far more appropriate for studying the evolution of SARS-CoV-2 viruses.
Findings illustrate the power of phylogenetic network analysis in the study of virus evolution
The study demonstrated that the alpha variant first reported in the U.K. could be a result of recombination of previously circulating variants of the virus.
The phylogenetic network analysis approach is currently being used to analyze the origin of other variants, including the ones that emerged in Brazil and India.
The recombinant origin of the alpha variant emphasizes the importance of achieving local as well as global control of the pandemic as fast as possible, as the rapid and continuous spread of the virus in any population facilitates the emergence of new variants.
Furthermore, these variants may have immune escape capabilities and start new waves of outbreaks, which might make it impossible to eliminate the SARS-CoV-2 virus from the human population entirely.
The study's findings illustrate the power and application of phylogenetic network analysis in the study of virus evolution.
According to the authors, phylogenetic network analysis can be used to study the evolutionary processes resulting in the emergence of new SARS-CoV-2 variants and several other viruses.
New vaccine production and distribution methods may help speed up worldwide production and delivery of COVID-19 vaccines effective against the recently emerged and emerging SARS-CoV-2 variants.
“Our study also demonstrated the power of phylogenetic network analysis and its competitive advantages over phylogenetic tree analysis in situations like the evolutionary study of the SARS-CoV-2 viruses.”
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Phylogenetic network analysis revealed the recombinant origin of the SARS-CoV-2 VOC202012/01 (B.1.1.7) variant first discovered in U.K., Xianfa Xie, Teash-Juan Lewis, Nikoli Green, Zhenping Wang, bioRxiv, 2021.06.24.449840; doi: https://doi.org/10.1101/2021.06.24.449840, https://www.biorxiv.org/content/10.1101/2021.06.24.449840v1
Posted in: Device / Technology News | Medical Research News | Disease/Infection News
Tags: Coronavirus, Coronavirus Disease COVID-19, Efficacy, Encephalitis, Evolution, Genome, HIV, Influenza, Pandemic, Public Health, Research, Respiratory, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Vaccine, Virus

Written by
Susha Cheriyedath
Susha has a Bachelor of Science (B.Sc.) degree in Chemistry and Master of Science (M.Sc) degree in Biochemistry from the University of Calicut, India. She always had a keen interest in medical and health science. As part of her masters degree, she specialized in Biochemistry, with an emphasis on Microbiology, Physiology, Biotechnology, and Nutrition. In her spare time, she loves to cook up a storm in the kitchen with her super-messy baking experiments.
Source: Read Full Article