The ongoing coronavirus disease 2019 (COVID-19) pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is still spreading across the world. Its spread is fueled by the emergence of newer, more infectious variants such as the B.1.1.7 variant, which emerged in the UK towards the latter part of 2020.
A new preprint on the medRxiv* server describes the impending rise to dominance of the B.1.1.7 variant in the US. Understanding this development will help to shape preventive action to reduce the infection and mortality rates.
The B.1.1.7 variant
The SARS-CoV-2 Variant of Concern (VOC) 202012/01 (a.k.a. 501Y.V1; B.1.1.7) lineage was first identified during the second half of 2020. Its early detection was due to large-scale and rapid sequencing of SARS-CoV-2 in the UK, as part of that country’s genomic surveillance program.
The UK variant shares the N501Y mutation with the South African and Brazil variants, but also has additional mutations. These ‘signature’ mutations include a deletion at 69/70, deletion at 144, along with A570D, D614G, P681H, T716I, S982A, and D1118H.
The UK variant is 40-70% more infectious than other variants, perhaps because the N501Y enhances the binding affinity of the SARS-CoV-2 spike protein with the host cell receptor, angiotensin-converting enzyme 2 (ACE2). In addition, it may cause up to 30% higher mortality rates.
The polymerase chain reaction (PCR) tests used to diagnose SARS-CoV-2 infection may offer an important clue to the presence of such viral lineages. This is because they have significantly different sequences at the target probe sites used in the test. The UK variant has a deletion at the 69/70 location, which typically leads to non-detection of the S gene by PCR tests based on the parent strains.
This is called S gene target failure (SGTF). In samples showing SGTF in the UK, the percentage of B.1.1.7 strains went up from 3% to over 90%, in the period beginning the week of October 12, 2020, and ending the week of November 30, 2020.
Increase in proportion of B.1.1.7 within SGTF
The current study explored the spread and prevalence of this variant in the USA. The researchers selected only SGTF samples for sequencing, as such samples are automatically enriched for B.1.1.7. The database comprised all SARS-CoV-2-positive samples tested at Helix facilities from July 2020 onwards, about half a million in all.
The Helix test for COVID-19 tests for three target antigens, the spike (S), the nucleocapsid (N), and the ORF1ab (open reading frame 1ab). If a sample detected both the N and ORF1ab but not the spike, it was termed an SGTF sample.
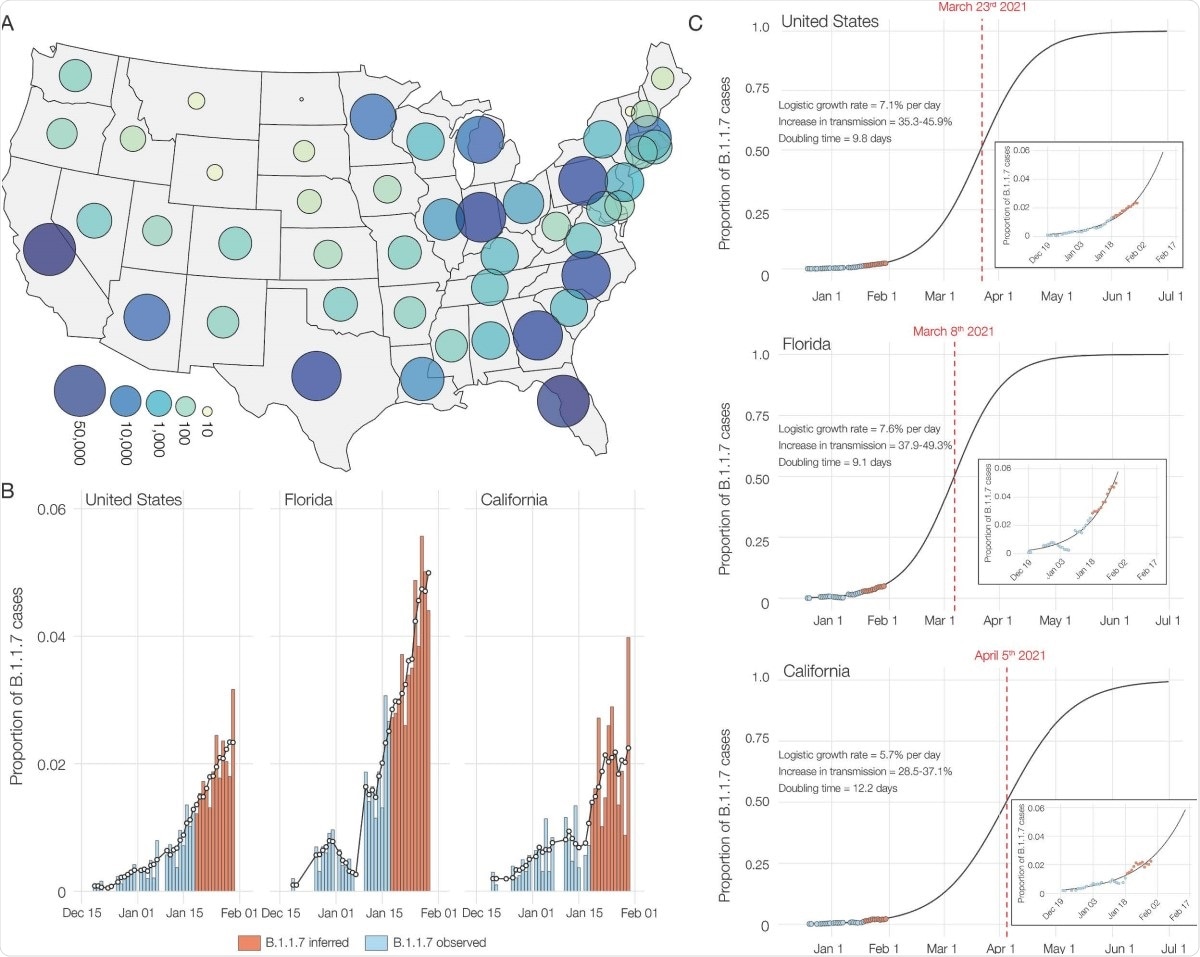
From the early part of October 2020, low-frequency SGTF began to be seen consistently, comprising 0.2% of daily positive tests by October 18. In January 2021, it rose from 0.8% to 4.2% from the first week to the last. Of all SGTF samples, 3.6% were of the B.1.17 lineage.
Also, SGTF samples were found in 22 of the 53 US states and territories in January 2021, comprising over 1% in many places, but with an uneven distribution.
Identification of B.1.1.7 by sequencing
Sequencing of the 460 SGTF samples obtained from December 2020 through January 2021, they found 45% to be of the B.1.1.7 variant across ten states. At least one case has been reported to be caused by this variant in 33 states and territories, altogether, using data from all testing laboratories.
In the current study, almost half came from California, and a slightly lesser number from Florida. These two states made up almost 180 of the B.1.1.7 sequences.
The Florida sequences most often contained an additional K1191N mutation as well as, in a single case, the immune escape mutation Q493K.
Increased growth rate of B.1.1.7 in the USA
The researchers also found that B.1.1.7 comprised 90% of all SGTF samples by the middle of January 2020, from a high of 95% in California to a low of 70% in Florida. By the end of the month, this variant caused 2% of all COVID-19 cases in the country, with an increase in transmissibility of 35-46%. The doubling time for this variant was estimated to be ~10 days in the US overall, ~12 days and ~9 days in California and Florida, respectively.
Multiple introductions
The researchers also found that most of the B.1.1.7 sequences in the US were in two major clades, introduced independently into California. Smaller clades represented eight other introductions, such as the GA clade, containing three sequences from Georgia. There were 19 single strains caused by still other independent introductions.
The earliest introduction was reflected by the CA1 clade, which initiated sustained community spread in California in November 2020. Since then, multiple clades have been repeatedly brought into the country up to the present time. The B.1.1.7 variant has also been spreading between states and territories since December 2020, at least.
What are the implications?
Though B.1.1.7 variants are now at a low proportion of the circulating viral variants in the USA, it is growing at a much higher rate (an increase of 35-45%) relative to other strains. In fact, its doubling time indicates that the number of infections caused by this variant is doubling every 10 days.
These findings are consistent with those from other countries, and given the current trajectory of B.1.1.7 in the US, it is almost certainly destined to become the dominant SARS-CoV-2 lineage by March, 2021 across many US states.”
More research will be required, such as a strong national surveillance program involving genomic sequencing, to understand the real prevalence of this variant in the country. Its introduction, directionality of spread, and origin, are still unknown. However, the introductions seem to be associated with high-travel periods, with over a million travelers crossing checkpoints per day.
These include the peak Thanksgiving season as well as the Christmas and New Year holiday periods. The origin would therefore seem to be an introduction via international travel, which spread within the community rapidly.
The B.1.1.7 variant now makes up 90% of SGTF samples and its increased growth rate makes it likely that it will soon be simple to monitor its spread by using SGTF percentages. However, increased genomic surveillance is essential to assess and follow new variants that are emerging, possibly with higher transmissibility and virulence.
Unless decisive and immediate public health action is taken, the increased transmission rate of these lineages and resultant higher effective reproduction number of SARS-CoV-2 will likely have devastating consequences to COVID-19 mortality and morbidity in the US in a few months, if decisive action is not immediately taken.”
*Important Notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Washington, N. L. et al. (2021). Genomic epidemiology identifies emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. medRxiv preprint. doi: https://doi.org/10.1101/2021.02.06.21251159, https://www.medrxiv.org/content/10.1101/2021.02.06.21251159v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News | Healthcare News
Tags: ACE2, Angiotensin, Angiotensin-Converting Enzyme 2, binding affinity, Cell, Coronavirus, Coronavirus Disease COVID-19, Enzyme, Epidemiology, Frequency, Gene, Genomic, Genomic Sequencing, Genomics, Helix, Mortality, Mutation, Pandemic, Polymerase, Polymerase Chain Reaction, Protein, Public Health, Receptor, Reproduction, Research, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome

Written by
Dr. Liji Thomas
Dr. Liji Thomas is an OB-GYN, who graduated from the Government Medical College, University of Calicut, Kerala, in 2001. Liji practiced as a full-time consultant in obstetrics/gynecology in a private hospital for a few years following her graduation. She has counseled hundreds of patients facing issues from pregnancy-related problems and infertility, and has been in charge of over 2,000 deliveries, striving always to achieve a normal delivery rather than operative.
Source: Read Full Article